Publications
Last publication: Computational and Structural Biotechnology Journal, 27, 127-136 (2025). https://doi.org/10.1016/j.csbj.2024.12.004
Our Team Members Publications
2024
|
PUR-GEN: A web server for automated generation of polyurethane fragment libraries Katarzyna Szleper, Mateusz Cebula, Oksana Kovalenko, Artur Góra, Agata Raczyńska ARTICLE Computational and Structural Biotechnology Journal, 27, 127-136 (2025). https://doi.org/10.1016/j.drudis.2024.104217 |
||||||||
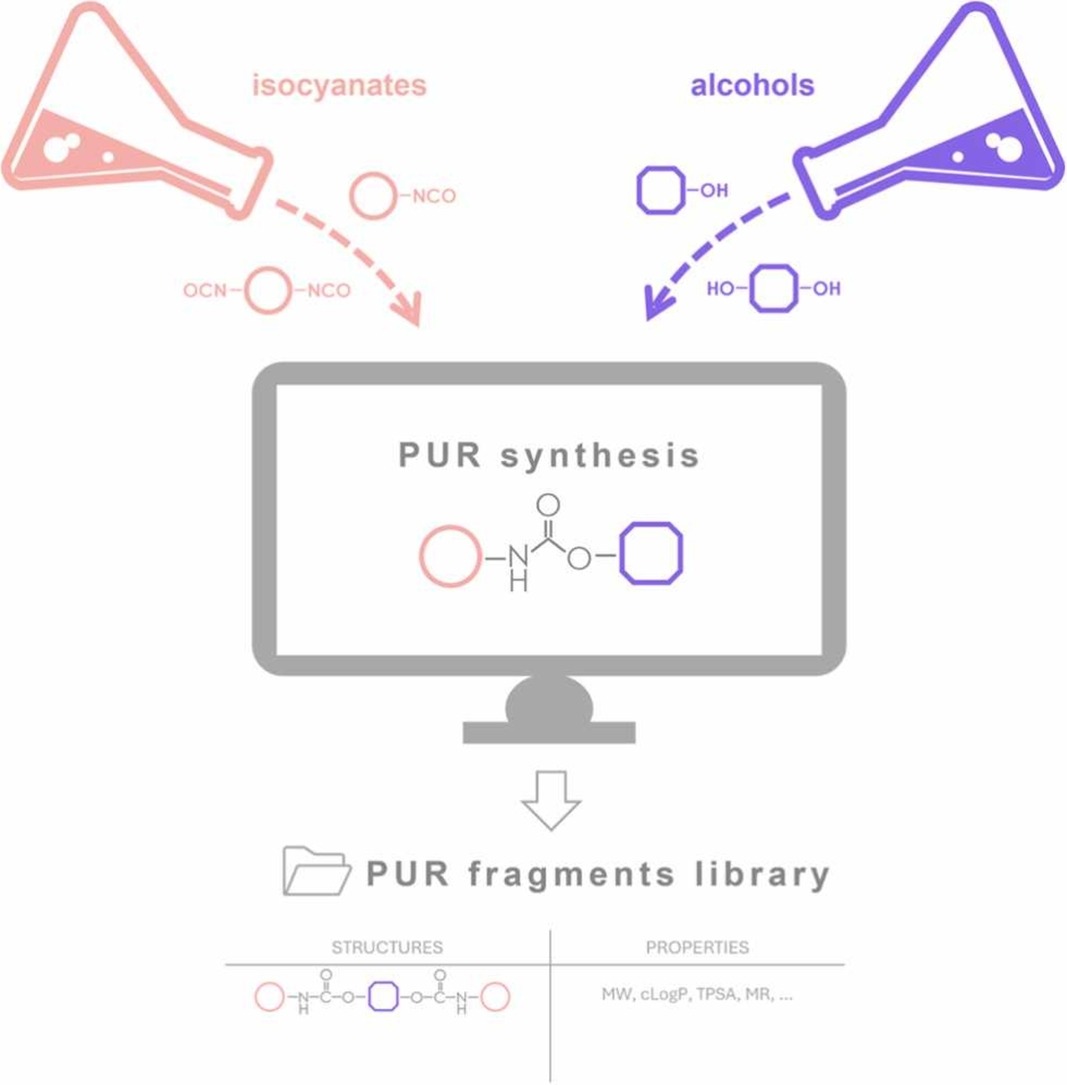
ABSTRACT
The biodegradation of synthetic polymers offers a promising solution for sustainable plastic recycling. Polyurethanes (PUR) stand out among these polymers due to their susceptibility to enzymatic hydrolysis. However, the intricate 3D structures formed by PUR chains present challenges for biodegradation studies, both computational and experimental. To facilitate in silico research, we introduce PUR-GEN, a web server tailored for the automated generation of PUR fragment libraries. PUR-GEN allows users to input isocyanate and alcohol structural units, facilitating the creation of combinatorial oligomer libraries enriched with conformers and compound property tables. PUR-GEN can serve as a valuable tool for designing PUR fragments to mimic PUR structure interactions with proteins, as well as characterising simplistic PUR models. To illustrate an application of the web server, we present a case study on selected four cutinases and three urethanases with experimentally confirmed PUR-degrading activity or ability to hydrolyse carbamates. The use of PUR-GEN in molecular docking of 414 generated oligomers provides an example of the pipeline for initiation of the PUR degrading enzymes discovery.
|
Water Migration through Enzyme Tunnels Is Sensitive to the Choice of Explicit Water Model Aravind Selvaram Thirunavukarasu, Katarzyna Szleper, Gamze Tanriver, Igor Marchlewski, Karolina Mitusinska, Artur Gora, Jan Brezovsky, ARTICLE J. Chem. Inf. Model. X, XXX (2024). https://doi.org/10.1021/acs.jcim.4c01177 |
||||||||
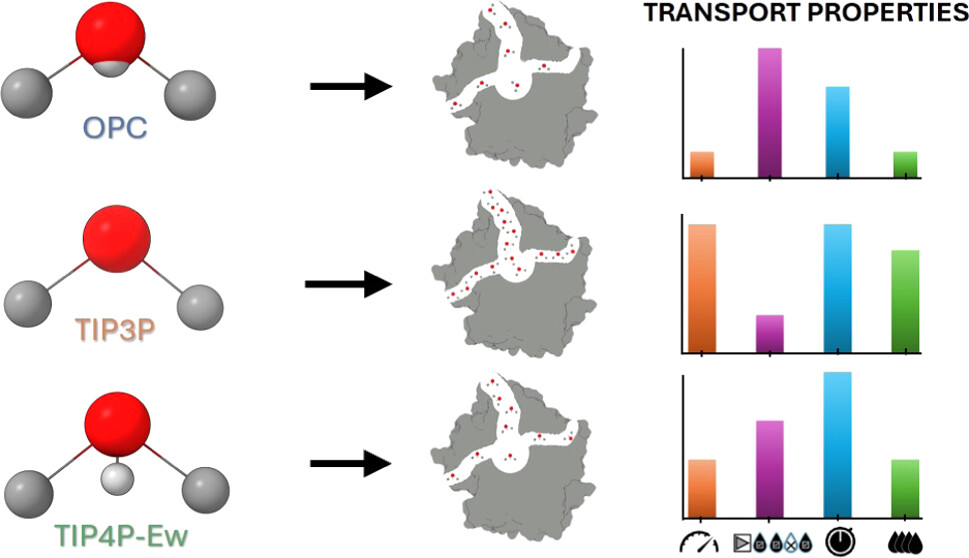
ABSTRACT
The utilization of tunnels and water transport within enzymes is crucial for their catalytic function as water molecules can stabilize bound substrates and help with unbinding processes of products and inhibitors. Since the choice of water models for molecular dynamics simulations was shown to determine the accuracy of various calculated properties of the bulk solvent and solvated proteins, we have investigated if and to what extent water transport through the enzyme tunnels depends on the selection of the water model. Here, we focused on simulating enzymes with various well-defined tunnel geometries. In a systematic investigation using haloalkane dehalogenase as a model system, we focused on the well-established TIP3P, OPC, and TIP4P-Ew water models to explore their impact on the use of tunnels for water molecule transport. The TIP3P water model showed significantly faster migration, resulting in the transport of approximately 2.5 times more water molecules compared to that of the OPC and 1.7 times greater than that of the TIP4P-Ew. Finally, the transport was 1.4-fold more pronounced in TIP4P-Ew than in OPC. The increase in migration of TIP3P water molecules was mainly due to faster transit times through dehalogenase tunnels. We observed similar behavior in two different enzymes with buried active sites and different tunnel network topologies, i.e., alditol oxidase and cytochrome P450, indicating that our findings are likely not restricted to a particular enzyme family. Overall, this study showcases the critical importance of water models in comprehending the use of enzyme tunnels for small molecule transport. Given the significant role of water availability in various stages of the catalytic cycle and the solvation of substrates, products, and drugs, choosing an appropriate water model may be crucial for accurate simulations of complex enzymatic reactions, rational enzyme design, and predicting drug residence times.
|
Comprehensive analysis of acetylcholinesterase inhibitor and reactivator complexes: implications for drug design and antidote development Weronika Bagrowska, Angelika Karasewicz, Artur Góra, ARTICLE Drug Discovery Today 29(12), 104217 (2024). https://doi.org/10.1016/j.drudis.2024.104217 |
||||||||
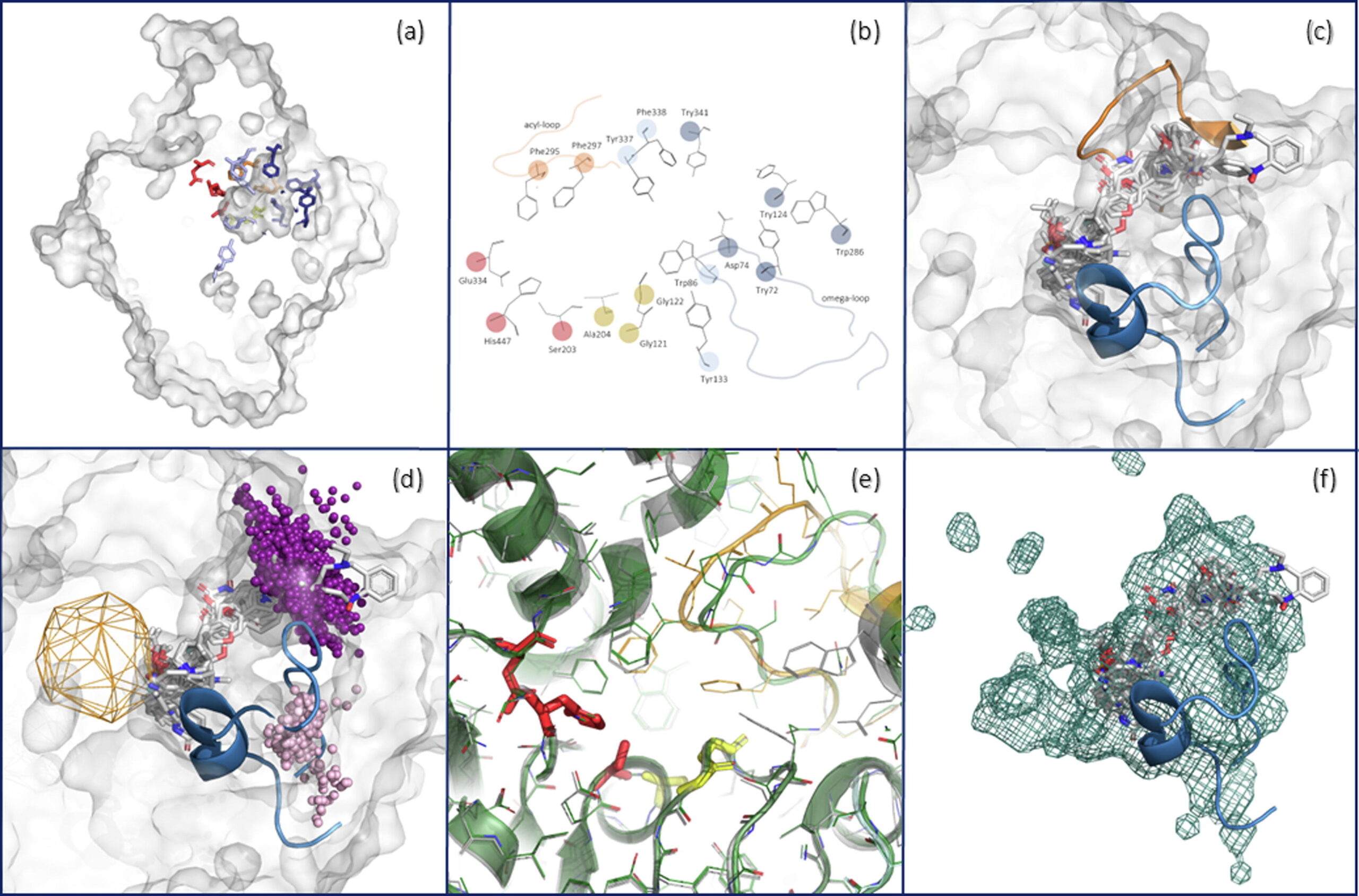
ABSTRACT
The main function of acetylcholinesterase (AChE) is to regulate the levels of one of the most important neurotransmitters: acetylcholine. This makes AChE an ideal molecular target for the treatment of neurodegenerative diseases and dementia (such as Alzheimer’s disease), as well as for the neutralisation of natural toxins (e.g., venom peptides) and chemical warfare agents. The significance of AChE inhibitors in slowing the progression of dementia, as well as the role of reactivators in treating poisoned individuals, is reflected in several co-crystallised complexes deposited in the Protein Data Bank. In this study, we analysed all deposited AChE–small-molecule complexes to gain insights into compound binding and to provide guidance for the future design of therapeutic drugs and new antidotes.
|
An overview on polyurethane-degrading enzymes Agata Raczyńska, Artur Góra, Isabelle André, ARTICLE Biotechnology Advances 77, 108439 (2024). https://doi.org/10.1016/j.biotechadv.2024.108439 |
||||||||
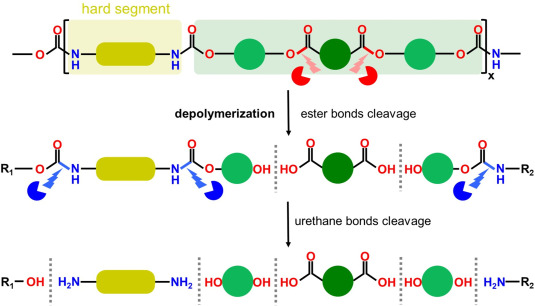
ABSTRACT
Polyurethanes (PUR) are durable synthetic polymers widely used in various industries, contributing significantly to global plastic consumption. PUR pose unique challenges in terms of degradability and recyclability, as they are characterised by intricate compositions and diverse formulations. Additives and proprietary structures used in commercial PUR formulations further complicate recycling efforts, making the effective management of PUR waste a daunting task.
In this review, we delve into the complex challenge of enzymatic degradation of PUR, focusing on the structural and functional attributes of both enzymes and PUR. We also present documented native enzymes with reported efficacy in hydrolysing specific bonds within PUR, analysis of these enzyme structures, reaction mechanisms, substrate specificity, and binding site architecture. Furthermore, we propose essential features for the future redesign of enzymes to optimise PUR biodegradation efficiency. By outlining prospective research directions aimed at advancing the field of enzymatic biodegradation of PUR, we aim to contribute to the development of sustainable solutions for managing PUR waste and reducing environmental pollution.
|
PBP-A, a cyanobacterial DD-peptidase with high specificity for amidated muropeptides, exhibits pH-dependent promiscuous activity harmful to Escherichia coli Gol Mohammad Dorrazehi, Matthias Winkle, Martin Desmet, Vincent Stroobant, Gamze Tanriver, Hervé Degand, Damien Evrard, Benoît Desguin, Pierre Morsomme, Jacob Biboy, Joe Gray, Karolina Mitusińska, Artur Góra, Waldemar Vollmer & Patrice Soumillion, ARTICLEOpen Access Scientific Reports 14, 13999 (2024). https://doi.org/10.1038/s41598-024-64806-x |
||||||||
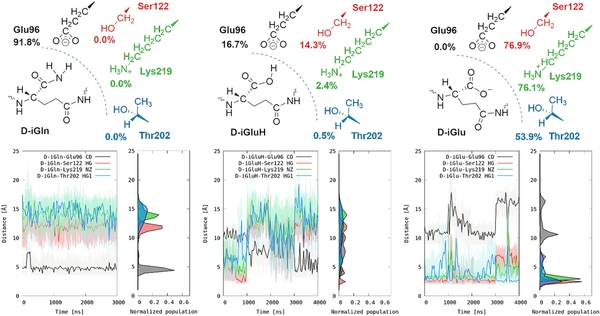
ABSTRACT
Penicillin binding proteins (PBPs) are involved in biosynthesis, remodeling and recycling of peptidoglycan (PG) in bacteria. PBP-A from Thermosynechococcus elongatus belongs to a cyanobacterial family of enzymes sharing close structural and phylogenetic proximity to class A β-lactamases. With the long-term aim of converting PBP-A into a β-lactamase by directed evolution, we simulated what may happen when an organism like Escherichia coli acquires such a new PBP and observed growth defect associated with the enzyme activity. To further explore the molecular origins of this harmful effect, we decided to characterize deeper the activity of PBP-A both in vitro and in vivo. We found that PBP-A is an enzyme endowed with DD-carboxypeptidase and DD-endopeptidase activities, featuring high specificity towards muropeptides amidated on the D-iso-glutamyl residue. We also show that a low promiscuous activity on non-amidated peptidoglycan deteriorates E. coli’s envelope, which is much higher under acidic conditions where substrate discrimination is mitigated. Besides expanding our knowledge of the biochemical activity of PBP-A, this work also highlights that promiscuity may depend on environmental conditions and how it may hinder rather than promote enzyme evolution in nature or in the laboratory.
|
Effects of γ-polyglutamic acid on grassland sandy soil properties and plant functional traits exposed to drought stress Tomasz Skalski, Ewelina Zając, Elżbieta Jędrszczyk, Katarzyna Papaj, Joanna Kohyt, Artur Góra, Anna Kasprzycka, Divine Shytum, Barbara Skowera, Agnieszka Ziernicka-Wojtaszek, ARTICLEOpen Access Scientific Reports 14, 3769 (2024). https://doi.org/10.1038/s41598-024-54459-1 |
||||||||
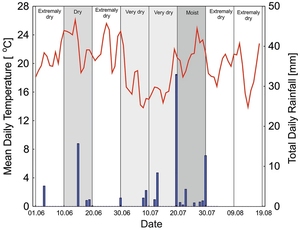
ABSTRACT
The current study provides field experimental data that support the use of γ-polyglutamic acid (γ-PGA) in drought stress and proposes its application in grassland management. We hypothesized that water treatment combined with PGA application to sandy soil would reduce drought stress in grasslands more effectively than watering alone. A randomized block design was used, with three replicate watering blocks (no watering, weekly watering, and monthly watering) and PGA treatments at four different concentrations (0%, 0.3%, 1%, and 2% PGA). The results showed that PGA acts as a biostimulant, alleviating the effects of stress in plants by: (1) increasing the availability of ions, especially K+, Zn2+, Mn2+, Fe2+/3+, Ca2+, and Mg2+, as well as N-NH4+, and N-NO3−, (2) elongating plant roots, (3) increasing the aboveground biomass, (4) improving the resprouting capacity of the dominant grass Nardus stricta, and (5) improving the regeneration of dicotyledons. In the case of meadows on sandy soils, the use of low PGA concentrations (0.3% or 1%) was the most beneficial for the availability of macro- and microelements and improving the functional traits of plants. Irrigation had a greater effect than using PGA only for the dicotyledon to monocotyledon ratio.
2023
|
Recent Advances in Studying Toll-like Receptors with the Use of Computational Methods Bzówka M, Bagrowska W, Góra A, ARTICLEOpen Access J. Chem. Inf. Model., 2023, 63, 12, 3669–3687. https://doi.org/10.1021/acs.jcim.3c00419 |
||||||||
ABSTRACT
Toll-like receptors (TLRs) are transmembrane proteins that recognize various molecular patterns and activate signaling that triggers the immune response. In this review, our goal is to summarize how, in recent years, various computational solutions have contributed to a better understanding of TLRs, regarding both their function and mechanism of action. We update the recent information about small-molecule modulators and expanded the topic toward next-generation vaccine design, as well as studies of the dynamic nature of TLRs. Also, we underline problems that remain unsolved.
|
Transient binding sites at the surface of haloalkane dehalogenase LinB as locations for fine-tuning enzymatic activity Raczyńska A, Kapica P, Papaj K, Stańczak A, Shyntum D, Spychalska P, Byczek-Wyrostek A, Góra A, ARTICLEOpen Access PLoS ONE 18(2), e0280776. https://doi.org/10.1371/journal.pone.0280776 |
||||||||
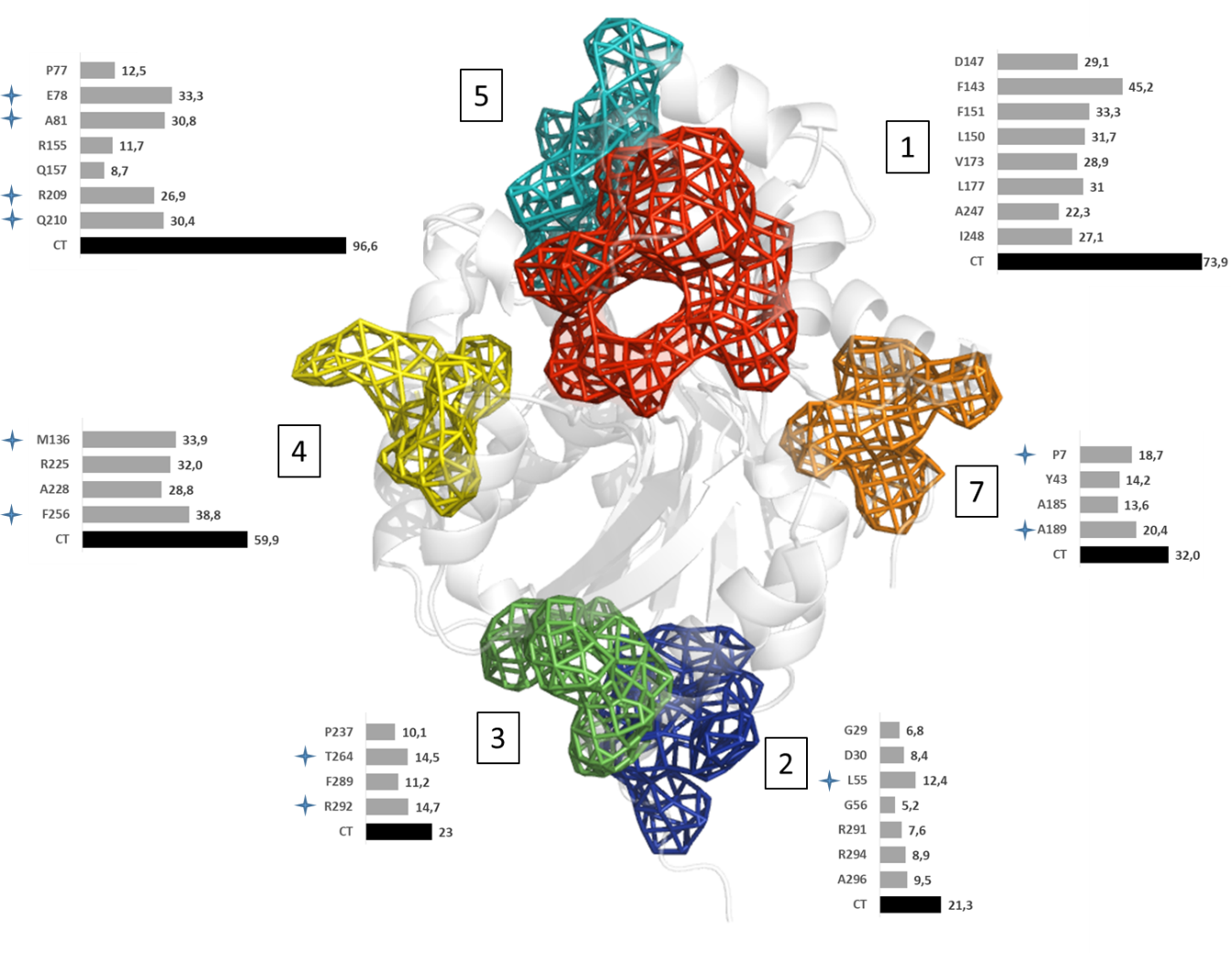
ABSTRACT
The haloalkane dehalogenase LinB is a well-known enzyme that contains buried active site and is used for many modelling studies. Using classical molecular dynamics simulations of enzymes and substrates, we searched for transient binding sites on the surface of the LinB protein by calculating maps of enzyme-ligand interactions that were then transformed into sparse matrices. All residues considered as functionally important for enzyme performance (e.g., tunnel entrances) were excluded from the analysis to concentrate rather on non-obvious surface residues. From a set of 130 surface residues, twenty-six were proposed as a promising improvement of enzyme performance. Eventually, based on rational selection and filtering out the potentially unstable mutants, a small library of ten mutants was proposed to validate the possibility of fine-tuning the LinB protein. Nearly half of the predicted mutant structures showed improved activity towards the selected substrates, which demonstrates that the proposed approach could be applied to identify non-obvious yet beneficial mutations for enzyme performance especially when obvious locations have already been explored.
2022
|
Evolution of tunnels in α/β-hydrolase fold proteins—What can we learn from studying epoxide hydrolases? Bzówka M, Mitusińska K, Raczyńska A, Skalski T, Samol A, Bagrowska W, Magdziarz T, Góra A, ARTICLEOpen Access PLOS Computational Biology 2022, 18 (5), e1010119. https://doi.org/10.1371/journal.pcbi.1010119 |
||||||||
ABSTRACT
The evolutionary variability of a protein’s residues is highly dependent on protein region and function. Solvent-exposed residues, excluding those at interaction interfaces, are more variable than buried residues whereas active site residues are considered to be conserved. The abovementioned rules apply also to α/β-hydrolase fold proteins—one of the oldest and the biggest superfamily of enzymes with buried active sites equipped with tunnels linking the reaction site with the exterior. We selected soluble epoxide hydrolases as representative of this family to conduct the first systematic study on the evolution of tunnels. We hypothesised that tunnels are lined by mostly conserved residues, and are equipped with a number of specific variable residues that are able to respond to evolutionary pressure. The hypothesis was confirmed, and we suggested a general and detailed way of the tunnels’ evolution analysis based on entropy values calculated for tunnels’ residues. We also found three different cases of entropy distribution among tunnel-lining residues. These observations can be applied for protein reengineering mimicking the natural evolution process. We propose a ‘perforation’ mechanism for new tunnels design via the merging of internal cavities or protein surface perforation. Based on the literature data, such a strategy of new tunnel design could significantly improve the enzyme’s performance and can be applied widely for enzymes with buried active sites.
|
Geometry-Based versus Small-Molecule Tracking Method for Tunnel Identification: Benefits and Pitfalls. Mitusińska K, Bzówka M, Magdziarz T, Góra A, ARTICLEOpen Access J. Chem. Inf. Model. 2022, 62, 24, 6803–6811. https://doi.org/10.1021/acs.jcim.2c00985 |
||||||||
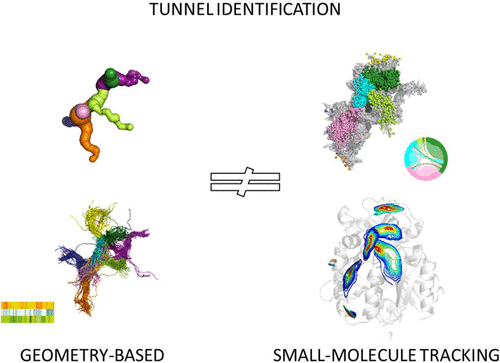
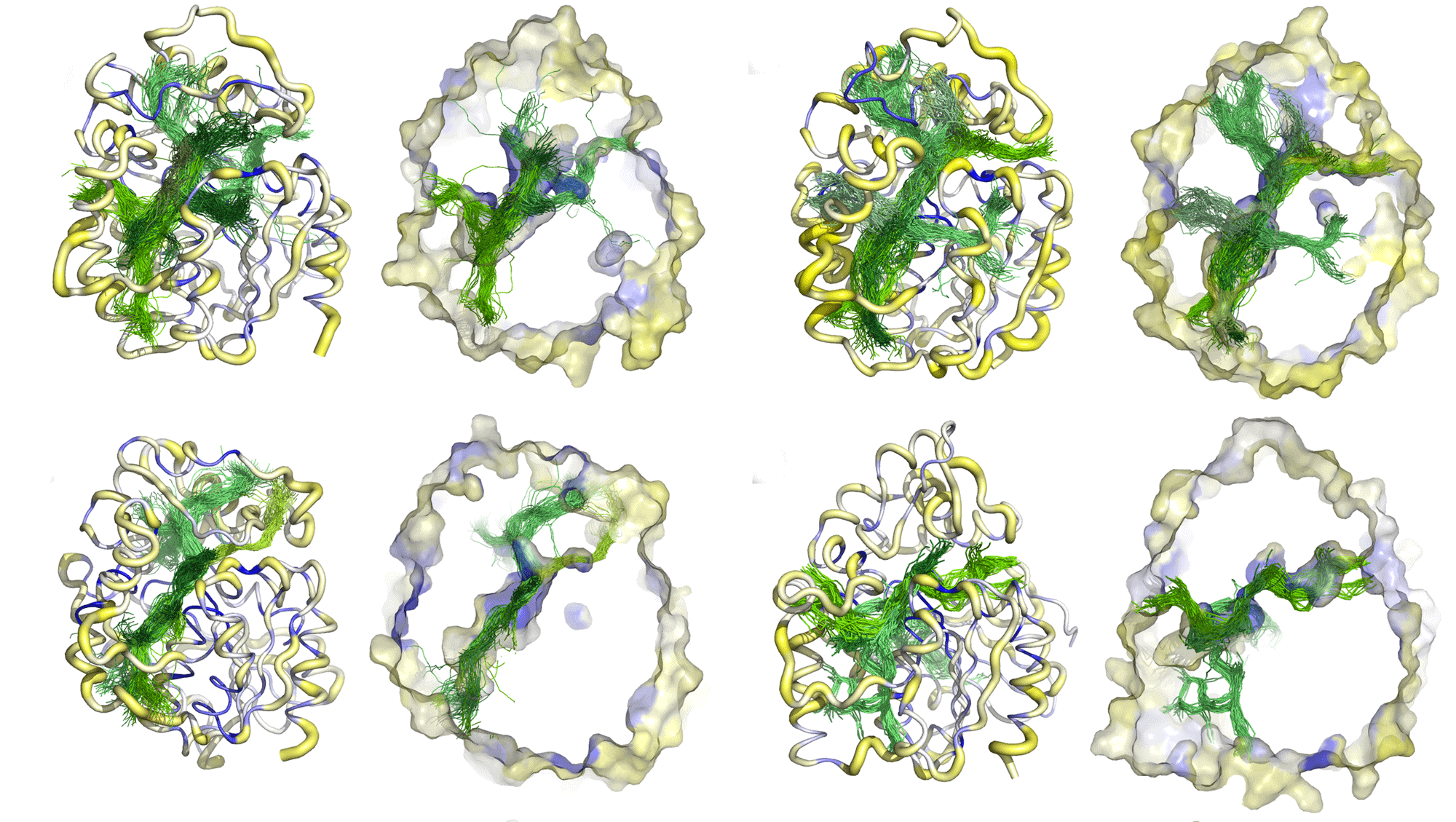
ABSTRACT
Different methods for tunnel identification, geometry-based and small-molecule tracking approaches, were compared to provide their benefits and pitfalls. Results obtained for both crystal structures and molecular dynamics (MD) simulations were analyzed to investigate if a more computationally demanding method would be beneficial. Careful examination of the results is essential for the low-diameter tunnel description, and assessment of the tunnel functionality based only on their geometrical parameters is challenging. We showed that the small-molecule tracking approach can provide a detailed description of the system; however, it can also be the most computationally demanding.
|
Structural Analysis of the Effect of Asn107Ser Mutation on Alg13 Activity and Alg13-Alg14 Complex Formation and Expanding the Phenotypic Variability of ALG13-CDG. Mitusińska K, Góra A, Bogdańska A, Rożdżyńska-Świątkowska A, Tylki-Szymańska A, Jezela-Stanek A ARTICLEOpen Access Biomolecules, 2022, 12(3) 398. https://doi.org/10.3390/biom12030398 |
||||||||
ABSTRACT
Congenital Disorders of Glycosylation (CDG) are multisystemic metabolic disorders showing highly heterogeneous clinical presentation, molecular etiology, and laboratory results. Here, we present different transferrin isoform patterns (obtained by isoelectric focusing) from three female patients harboring the ALG13 c.320A>G mutation. Contrary to other known variants of type I CDGs, where transferrin isoelectric focusing revealed notably increased asialo- and disialotransferrin fractions, a normal glycosylation pattern was observed in the probands. To verify this data and give novel insight into this variant, we modeled the human Alg13 protein and analyzed the dynamics of the apo structure and the complex with the UDP-GlcNAc substrate. We also modeled the Alg13-Alg14 heterodimer and ran multiple simulations of the complex in the presence of the substrate. Finally, we proposed a plausible complex formation mechanism.
|
Evaluation of Xa inhibitors as potential inhibitors of the SARS-CoV-2 Mpro protease. Papaj K, Spychalska P, Kapica P, Fisher A, Nowak J, Bzówka M, Sellner M, Lill MA, Smieško M, Góra A ARTICLEOpen Access PloS ONE 2022, 17(1):e0262482. https://doi.org/10.1371/journal.pone.0262482 |
||||||||
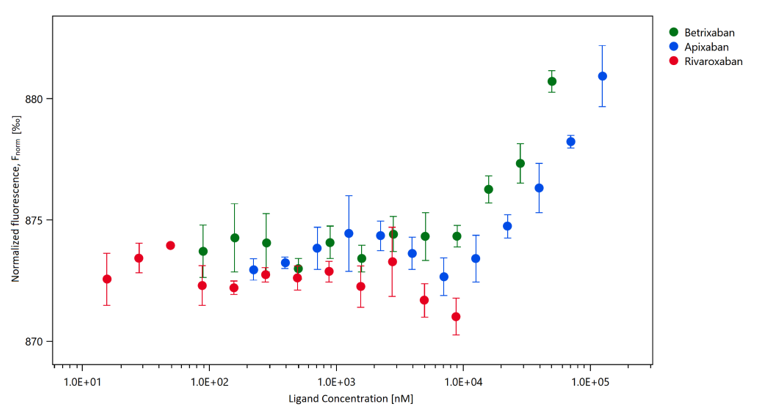
ABSTRACT
Based on previous large-scale in silico screening several factor Xa inhibitors were proposed to potentially inhibit SARS-CoV-2 Mpro. In addition to their known anticoagulants activity this potential inhibition could have an additional therapeutic effect on patients with COVID-19 disease. In this study we examined the binding of the Apixaban, Betrixaban and Rivaroxaban to the SARS-CoV-2 Mpro with the use of the MicroScale Thermophoresis technique. Our results indicate that the experimentally measured binding affinity is weak and the therapeutic effect due to the SARS-CoV-2 Mpro inhibition is rather negligible.
|
Structure-function relationship between soluble epoxide hydrolases structure and their tunnel network. Mitusińka K, Wojsa P, Bzówka M, Raczyńcsla A, Bagrowska W, Samol A, Kapica P, Góra A ARTICLEOpen Access Computational and Structural Biotechnology Journal 20, 2022, 193-205. https://doi.org/10.1016/j.csbj.2021.10.042 |
||||||||
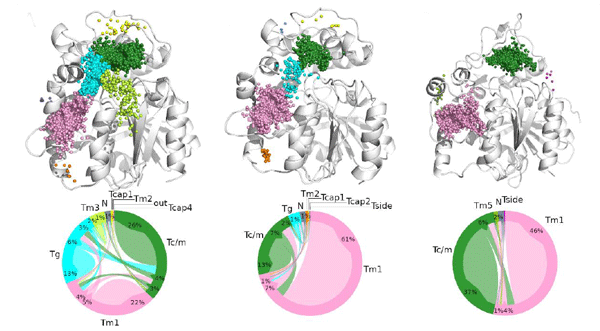
ABSTRACT
Enzymes with buried active sites maintain their catalytic function via a single tunnel or tunnel network. In this study we analyzed the functionality of soluble epoxide hydrolases (sEHs) tunnel network, by comparing the overall enzyme structure with the tunnel’s shape and size. sEHs were divided into three groups based on their structure and the tunnel usage. The obtained results were compared with known substrate preferences of the studied enzymes, as well as reported in our other work evolutionary analyses data. The tunnel network architecture corresponded well with the evolutionary lineage of the source organism and large differences between enzymes were observed from long fragments insertions. This strategy can be used during protein re-engineering process for large changes introduction, whereas tunnel modification can be applied for fine-tuning of enzyme.
2021
|
Investigation of Thiocarbamates as Potential Inhibitors of the SARS-CoV-2 Mpro. Papaj K, Spychalska P, Hopko K, Kapica P, Fisher A, Lill MA, Bagrowska W, Nowak J, Szleper K, Smieško M, Kasprzycka A, Góra A ARTICLEOpen Access Pharmaceuticals 2021, 14(11), 1153. https://doi.org/10.3390/ph14111153 |
||||||||
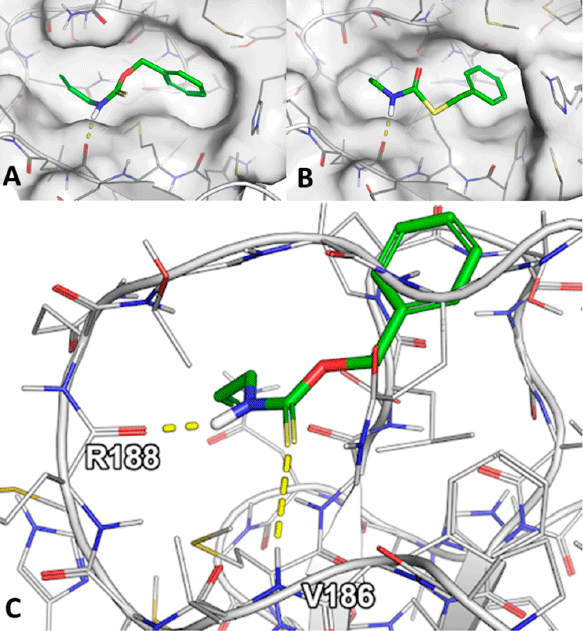
ABSTRACT
In the present study we tested, using the microscale thermophoresis technique, a small library of thionocarbamates, thiolocarbamates, sulfide and disulfide as potential lead compounds for SARS-CoV-2 Mpro drug design. The successfully identified binder is a representative of the thionocarbamates group with a high potential for future modifications aiming for higher affinity and solubility. The experimental analysis was extended by computational studies that show insufficient accuracy of the simplest and widely applied approaches and underline the necessity of applying more advanced methods to properly evaluate the affinity of potential SARS-CoV-2 Mpro binders.
|
Computational insights into the known inhibitors of human soluble epoxide hydrolase. Bzówka M., Mitusińska K., Hopko K., Góra A. ARTICLEOpen Access Drug Discovery Today, 26(8):1914-1921. https://doi.org/10.1016/j.drudis.2021.05.017 |
||||||||
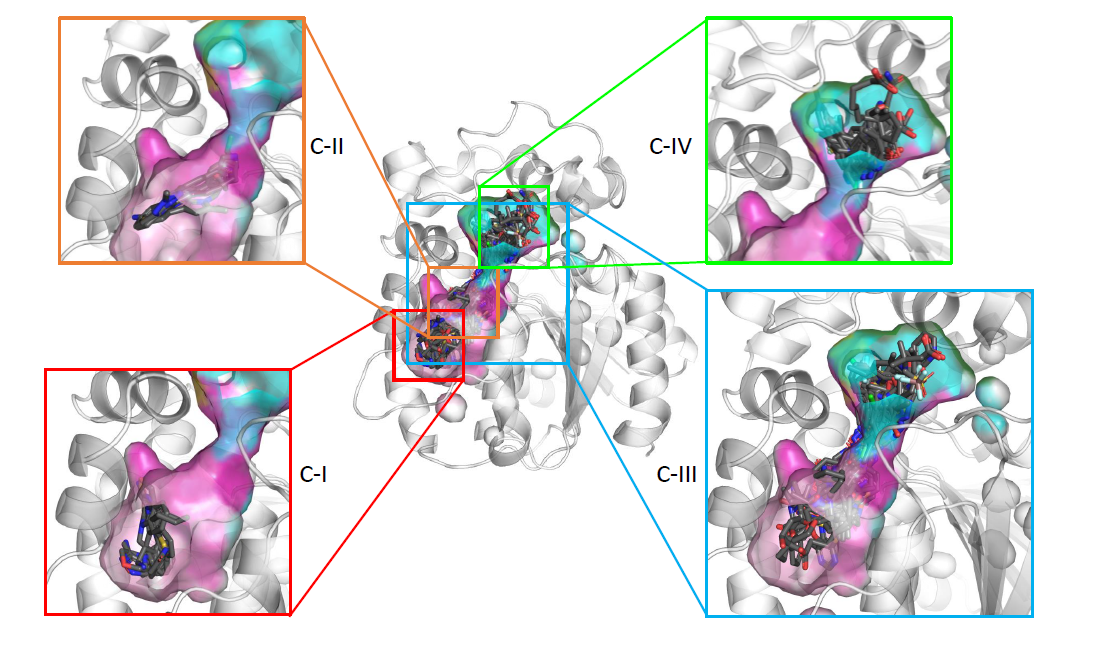
ABSTRACT
Human soluble epoxide hydrolase (hsEH) is involved in the hydrolysis of epoxyeicosatrienoic acids (EETs), which have potent anti-inflammatory properties. Given that EET conversion generates nonbioactive molecules, inhibition of this enzyme would be beneficial. Past decades of work on hsEH inhibitors resulted in numerous potential compounds, of which a hundred hsEH–ligand complexes were crystallized and deposited in the Protein Data Bank (PDB). We analyzed all deposited hsEH–ligand complexes to gain insight into the binding of inhibitors and to provide feedback on the future drug design processes. We also reviewed computationally driven strategies that were used to propose novel hsEH inhibitors.
|
AQUA-DUCT: Analysis of Molecular Dynamics Simulations of Macromolecules with the Use of Molecular Probes [Article v1.0]. Mitusińska K., Raczyńska A., Wojsa P., Bzówka M., Góra A. ARTICLEOpen Access Living Journal of Computational Molecular Science 2 (1), 21383 https://doi.org/10.33011/livecoms.2.1.21383 |
||||||||
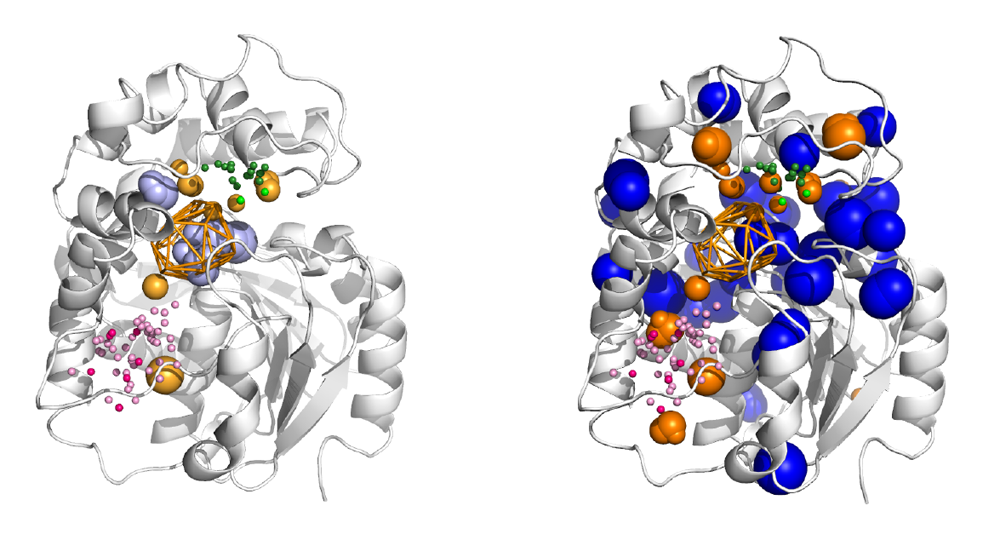
ABSTRACT
AQUA-DUCT software reverses the standard approach of the molecular dynamics simulations analysis of macromolecules, focusing on solvent, cosolvent and small ligands analysis considered as speci c molecular probes instead of analysis of macromolecules atoms movement. Here we present six basic tutorials instructing the users in the best practices for preparing, carrying out, and analysing AQUA-DUCT results in various applications. Users are expected to already have signi cant experience with running standard molecular dynamics simulations in any dedicated software (e.g., Amber, GROMACS, NAMD), and usage of PyMOL visualising software. The tutorials range from a basic analysis of multiple solvents trajectory used for identi cation of the entries/exits to the protein core, to a complex one, like identi cation of the key hot-spots in cosolvent MD simulations that can be used as an insight for macromolecules description, analysis, re-engineering, and for drug design.
|
Computational Selectivity Assessment of Protease Inhibitors against SARS-CoV-2. Fisher A., Sellner M., Mitusińska K., Bzówka M., Lill M.A., Góra A., Smeisko M. ARTICLEOpen Access International journal of molecular sciences 22 (4), 2065 https://doi.org/10.3390/ijms22042065 |
||||||||
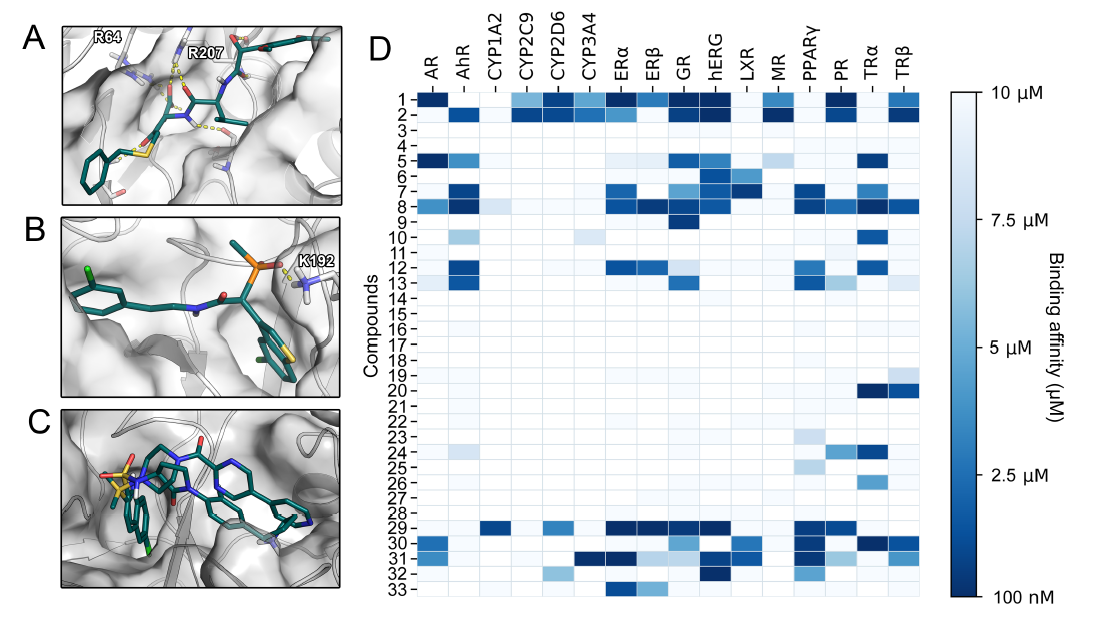
ABSTRACT
The pandemic of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) poses a serious global health threat. Since no specific therapeutics are available, researchers around the world screened compounds to inhibit various molecular targets of SARS-CoV-2 including its main protease (Mpro) essential for viral replication. Due to the high urgency of these discovery efforts, off-target binding, which is one of the major reasons for drug-induced toxicity and safety-related drug attrition, was neglected. Here, we used molecular docking, toxicity profiling, and multiple molecular dynamics (MD) protocols to assess the selectivity of 33 reported non-covalent inhibitors of SARSCoV-2 Mpro against eight proteases and 16 anti-targets. The panel of proteases included SARS-CoV Mpro, cathepsin G, caspase-3, ubiquitin carboxy-terminal hydrolase L1 (UCHL1), thrombin, factor Xa, chymase, and prostasin. Several of the assessed compounds presented considerable off-target binding towards the panel of proteases, as well as the selected anti-targets. Our results further suggest a high risk of off-target binding to chymase and cathepsin G. Thus, in future discovery projects, experimental selectivity assessment should be directed toward these proteases. A systematic selectivity assessment of SARS-CoV-2 Mpro inhibitors, as we report it, was not previously conducted.
2020
|
Structural and Evolutionary Analysis Indicate that the SARS-CoV-2 Mpro is a Challenging Target for Small-Molecule Inhibitors Design. Bzówka M., Mitusińska K., Raczyńska A., Samol A., Tuszyński J.A., Góra A. ARTICLEOpen Access International journal of molecular sciences 21 (9), 3099 https://doi.org/10.3390/ijms21093099 |
||||||||
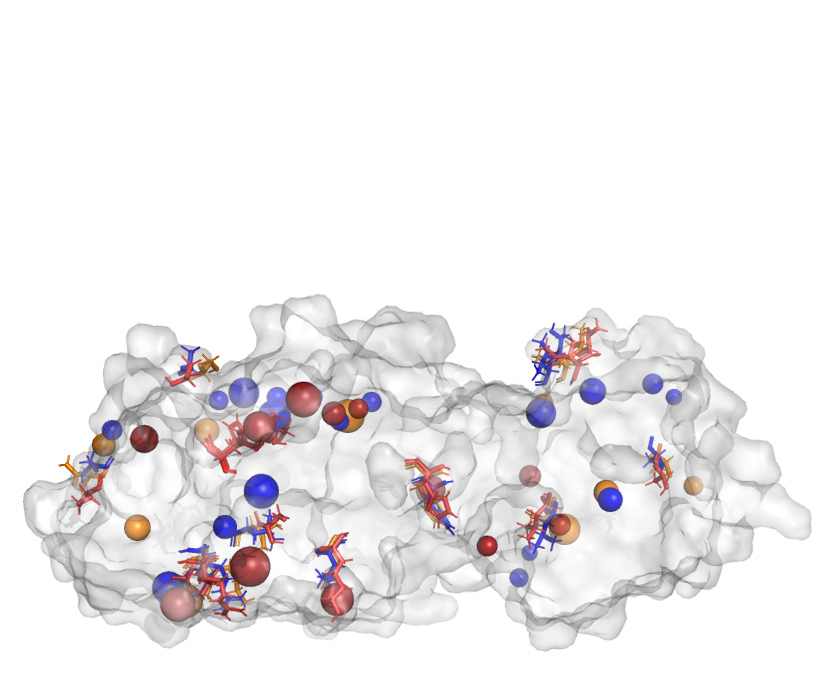
ABSTRACT
The novel coronavirus whose outbreak took place in December 2019 continues to spread at a rapid rate worldwide. In the absence of an effective vaccine, inhibitor repurposing or de novo drug design may offer a longer-term strategy to combat this and future infections due to similar viruses. Here, we report on detailed classical and mix-solvent molecular dynamics simulations of the main protease (Mpro) enriched by evolutionary and stability analysis of the protein. The results were compared with those for a highly similar SARS Mpro protein. In spite of a high level of sequence similarity, the active sites in both proteins show major differences in both shape and size indicating that repurposing SARS drugs for COVID-19 may be futile. Furthermore, analysis of the binding site’s conformational changes during the simulation time indicates its flexibility and plasticity, which dashes hopes for rapid and reliable drug design. Conversely, structural stability of the protein with respect to flexible loop mutations indicates that the virus’ mutability will pose a further challenge to the rational design of small-molecule inhibitors. However, few residues contribute significantly to the protein stability and thus can be considered as key anchoring residues for Mpro inhibitor design.
|
Simple Selection Procedure to Distinguish between Static and Flexible Loops. Mitusińska K., Skalski T., Góra A., Int. J. Mol. Sci. 2020, 21(7), 2293;ARTICLEOpen Access International journal of molecular sciences 21 (7), 2293 https://doi.org/10.3390/ijms21072293 |
||||||||
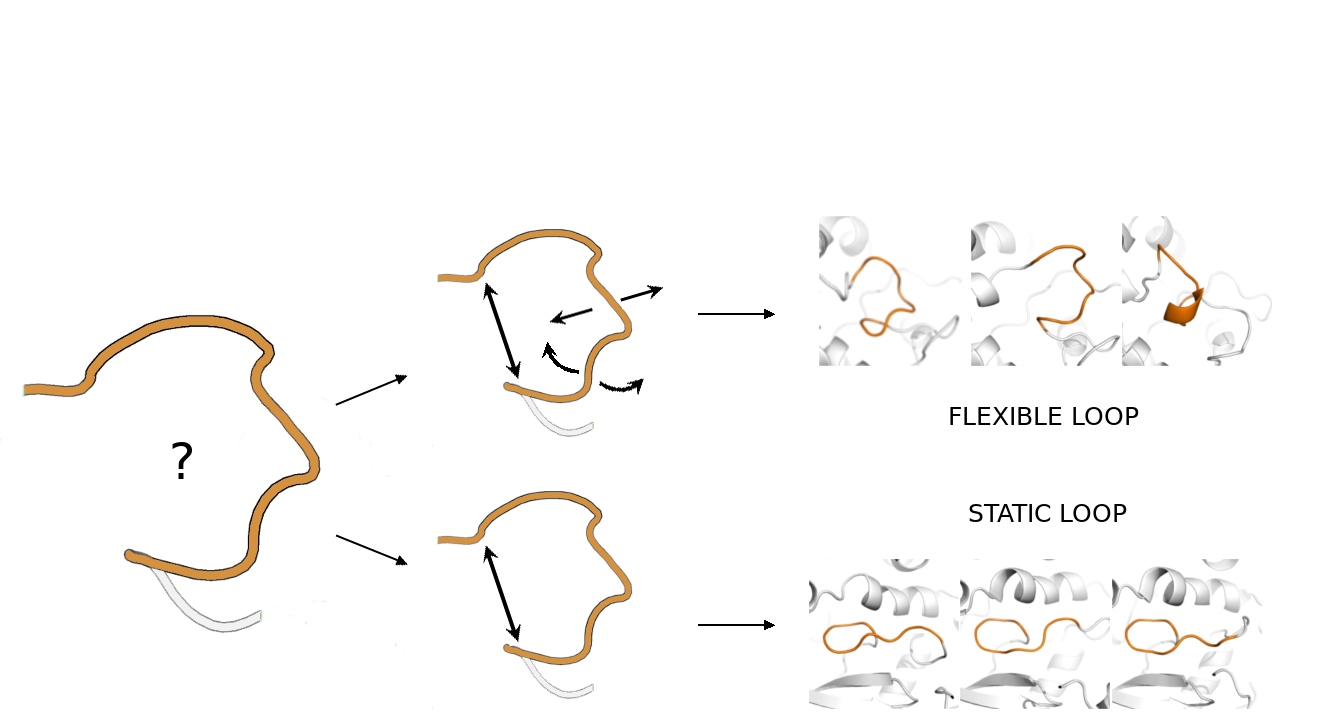
ABSTRACT
Loops are the most variable and unorganized elements of the secondary structure of proteins. Their ability to shift their shape can play a role in the binding of small ligands, enzymatic catalysis, or protein–protein interactions. Due to the loop flexibility, the positions of their residues in solved structures show the largest B-factors, or in a worst-case scenario can be unknown. Based on the loops’ movements’ timeline, they can be divided into slow (static) and fast (flexible). Although most of the loops that are missing in experimental structures belong to the flexible loops group, the computational tools for loop reconstruction use a set of static loop conformations to predict the missing part of the structure and evaluate the model. We believe that these two loop types can adopt different conformations and that using scoring functions appropriate for static loops is not sufficient for flexible loops. We showed that common model evaluation methods, are insufficient in the case of flexible solvent-exposed loops. Instead, we recommend using the potential energy to evaluate such loop models. We provide a novel model selection method based on a set of geometrical parameters to distinguish between flexible and static loops without the use of molecular dynamics simulations. We have also pointed out the importance of water network and interactions with the solvent for the flexible loop modelling.
|
Applications of water molecules for analysis of macromolecule properties. Mitusińska K., Raczyńska A., Bzówka M., Bagrowska W., Góra A. Computational and Structural Biotechnology Journal 2020, 18, 355-365 https://doi:10.1016/j.csbj.2020.02.001 ARTICLEOpen Access |
||||||||
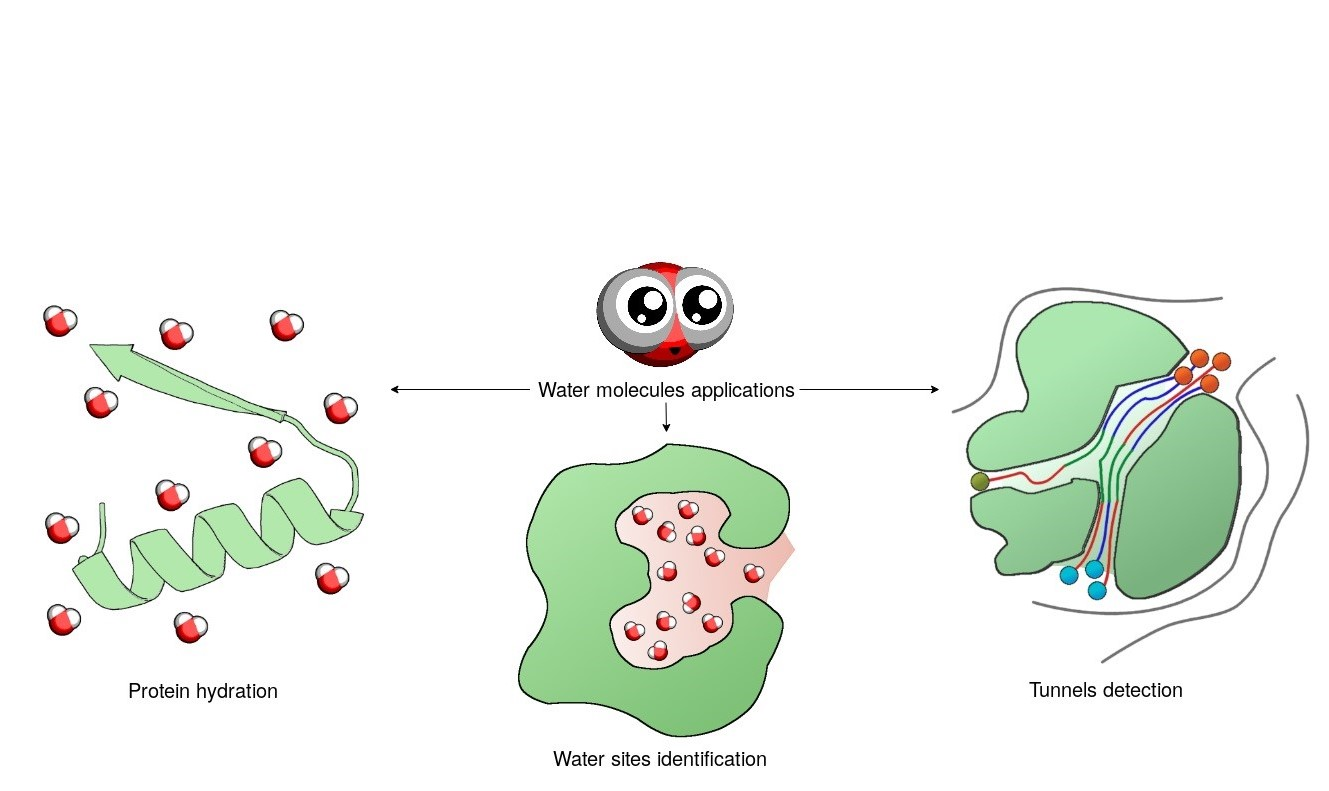
ABSTRACT
Water molecules maintain proteins’ structures, functions, stabilities and dynamics. They can occupy certain positions or pass quickly via a protein’s interior. Regardless of their behaviour, water molecules can be used for the analysis of proteins’ structural features and biochemical properties. Here, we present a list of several software programs that use the information provided by water molecules to: i) analyse protein structures and provide the optimal positions of water molecules for protein hydration, ii) identify high-occupancy water sites in order to analyse ligand binding modes, and iii) detect and describe tunnels and cavities. The analysis of water molecules’ distribution and trajectories sheds a light on proteins’ interactions with small molecules, on the dynamics of tunnels and cavities, on protein composition and also on the functionality, transportation network and location of functionally relevant residues. Finally, the correct placement of water molecules in protein crystal structures can significantly improve the reliability of molecular dynamics simulations.
|
Structure–bioavailability relationship study of genistein derivatives with antiproliferative activity on human cancer cell. Papaj, K.; Kasprzycka, A.; Góra, A.; Grajoszek, A.; Rzepecka, G.; Stojko, J.; Barski, J.-J.; Szeja, W.; Rusin, A. J Pharm Biomed Anal 2020, 185, 113216;https:// doi:10.1016/J.JPBA.2020.113216 ARTICLEOpen Access |
||||||||
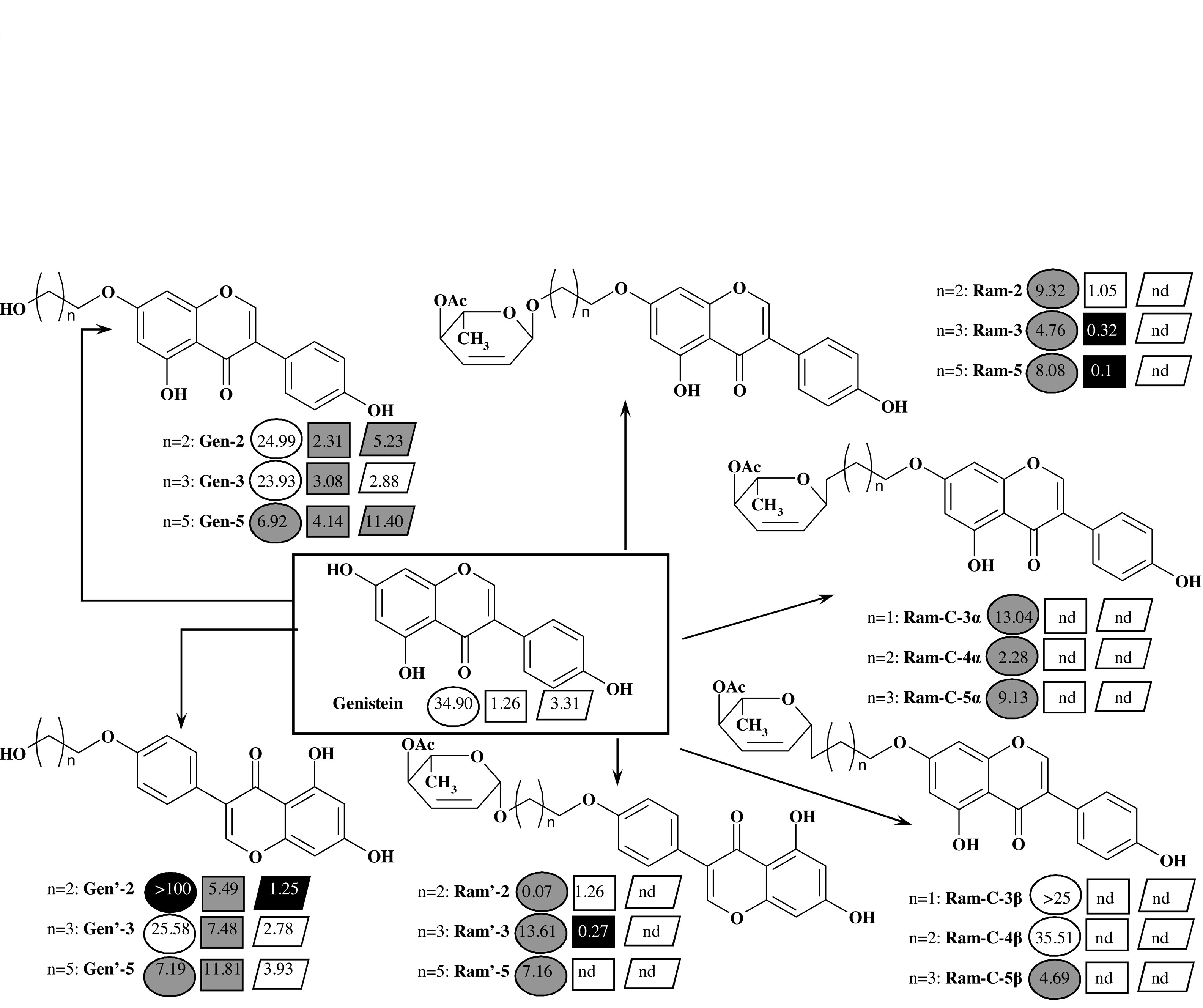
ABSTRACT
The present study assesses the in vitro and in vivo bioavailability of genistein derivatives, hydroxyalkyl- and glycosyl alkyl ethers (glycoconjugates). Studies were carried out using compounds that exhibit higher in vitro antiproliferative activity in comparison with the parent isoflavone. Based on in vitro experiments using the Parallel Artificial Membrane Permeability Assay (PAMPA) and the Caco-2 cell monolayer permeability model, we found that modification of the isoflavone structure by O-alkylation improved bioavailability in comparison to genistein. Additionally, the structure of the substituent and its position on genistein influenced the type of mechanism involved in the transport of compounds through biological membranes. The PAMPA assay showed that the structure of glycoconjugates had a significant influence on the passive transport of the genistein synthetic derivatives through a biological membrane. Preferentially the glycoconjugates containing O-glycosidic bond were transported and the transport rate decreased as the carbon linker increased. For glycoconjugates, determination of their transport and metabolism through the Caco-2 membrane was not possible due to interaction with the membrane surface, probably by the change of compound structure caused by contact with the cells or degradation in medium. The intestinal absorption and metabolism of genistein and three derivatives, Ram-3, Ram′-3 and Ram-C-4α (Fig. 1), were tested in vivo in rats. We found that in comparison to genistein, glycoconjugates were metabolized more slowly and to a lesser extent. As part of the in vivo research, we performed analysis of compound levels in plasma samples after enzymatic hydrolysis, but in the collected samples, analytes were not observed. We hypothesize that glycoconjugates compounds bind plasma proteins and were removed from the sample. In conclusion, we show that O-functionalization of the natural, biologically active isoflavone genistein can affect biological activity, bioavailability, and the rate of compound metabolism. The position of the substituent, the length of the linker and the structure of sugar moieties provides a tool for the optimization of the derivative’s biological properties.
2019
|
AQUA-DUCT 1.0: structural and functional analysis of macromolecules from an intramolecular voids perspective. Magdziarz T., Mitusińska K., Bzówka M., Raczyńska A., Stańczak A., Banas M., Bagrowska W., Góra A. ,Bioinformatics 2019, 1–3. https://doi.org/10.1093/bioinformatics/btz946 ARTICLEOpen Access |
||||||||
ABSTRACT
Tunnels, pores, channels, pockets and cavities contribute to proteins architecture and performance. However, analysis and characteristics of transportation pathways and internal binding cavities are performed separately. We aimed to provide universal tool for analysis of proteins integral interior with access to detailed information on the ligands transportation phenomena and binding preferences.
|
Can a Mononuclear Iron(III)-Superoxo Active Site Catalyze the Decarboxylation of Dodecanoic Acid in UndA to Produce Biofuels? Lin YT, Stańczak A, Manchev Y, Straganz GD, de Visser SP. Chemistry. 2019 Oct 4. https://doi.org/10.1002/chem.201903783 ARTICLEOpen Access |
||||||||
ABSTRACT
Decarboxylation of fatty acids is an important reaction in cell metabolism, but also has potential in biotechnology for the biosynthesis of hydrocarbons as biofuels. The recently discovered nonheme iron decarboxylase UndA is involved in the biosynthesis of 1-undecene from dodecanoic acid and using X-ray crystallography was assigned to be a mononuclear iron species. However, the work was contradicted by spectroscopic studies that suggested UndA to be more likely a dinuclear iron system. To resolve this controversy we decided to pursue a computational study on the reaction mechanism of fatty acid decarboxylation by UndA using iron(III)-superoxo and diiron(IV)-dioxo models. We tested several models with different protonation states of active site residues. Overall, however, the calculations imply that mononuclear iron(III)-superoxo is a sluggish oxidant of hydrogen atom abstraction reactions in UndA and will not be able to activate fatty acid residues by decarboxylation at room temperature. By contrast, a diiron-dioxo complex reacts with much lower hydrogen atom abstraction barriers and hence is a more likely oxidant in UndA.
|
Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase. Subramanian, K.; Mitusińska, K.; Raedts, J.; Almourfi, F.; Joosten, H.-J.; Hendriks, S.; Sedelnikova, S.E.; Kengen, S.W.M.; Hagen, W.R.; Góra, A.; Martins dos Santos, V.A.P.; Baker, P.J.; van der Oost, J.; Schaap, P.J., Biomolecules 2019, 9, 212. https://doi.org/10.3390/biom9060212 ARTICLEOpen Access |
||||||||
ABSTRACT
The cupin-type phosphoglucose isomerase (PfPGI) from the hyperthermophilic archaeon Pyrococcus furiosus catalyzes the reversible isomerization of glucose-6-phosphate to fructose-6-phosphate. We investigated PfPGI using protein-engineering bioinformatics tools to select functionally-important residues based on correlated mutation analyses. A pair of amino acids in the periphery of PfPGI was found to be the dominant co-evolving mutation. The position of these selected residues was found to be non-obvious to conventional protein engineering methods. We designed a small smart library of variants by substituting the co-evolved pair and screened their biochemical activity, which revealed their functional relevance. Four mutants were further selected from the library for purification, measurement of their specific activity, crystal structure determination, and metal cofactor coordination analysis. Though the mutant structures and metal cofactor coordination were strikingly similar, variations in their activity correlated with their fine-tuned dynamics and solvent access regulation. Alternative, small smart libraries for enzyme optimization are suggested by our approach, which is able to identify non-obvious yet beneficial mutations.
|
Reaction mechanism between Cu(II)-enolate complex and O2 as a test case for methodology used in DFT computational studies. A. Stańczak, A.Miłaczewska, T. Borowski, T. J Mol Model (2019) 25: 122. https://doi.org/10.1007/s00894-019-3998-3 ARTICLEOpen Access |
||||||||
ABSTRACT
The reaction mechanism of an intricate oxidation reaction of chlorodiketonate ligand of mononuclear Cu(II) complex was studied computationally employing five different models that differ in: a) basis set, b) the way that solvent corrections are included, and c) DFT functional. Qualitative and quantitative comparison of structures and enthalpy reaction profiles enabled us to assess how sensitive they are to the changes in computational methodology.
2018
|
Exploring Solanum Tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. K. Mitusińska, T. Magdziarz, M. Bzówka, A. Stańczak, A. Góra, Biomolecules 2018, 8(4): 143. DOI: https://doi.org/10.3390/biom8040143 ARTICLEOpen Access |
||||||||
ABSTRACT
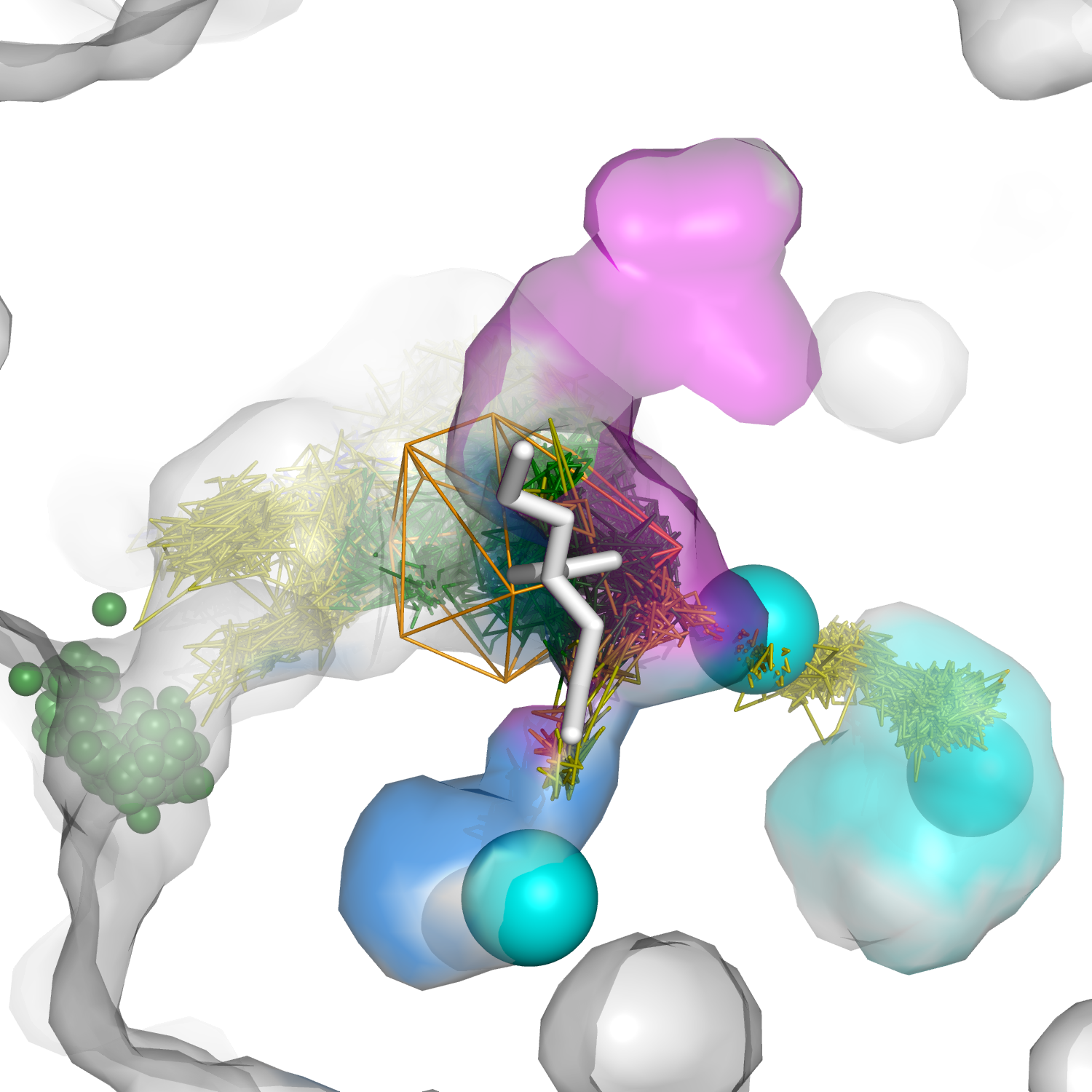
Several different approaches are used to describe the role of protein compartments and residues in catalysis and to identify key residues suitable for the modification of the activity or selectivity of the desired enzyme. In our research, we applied a combination of molecular dynamics simulations and a water tracking approach to describe the water accessible volume of Solanum tuberosum epoxide hydrolase. Using water as a molecular probe, we were able to identify small cavities linked with the active site: (i) one made up of conserved amino acids and indispensable for the proper positioning of catalytic water and (ii) two others in which modification can potentially contribute to enzyme selectivity and activity. Additionally, we identified regions suitable for de novo tunnel design that could also modify the catalytic properties of the enzyme. The identified hot-spots extend the list of the previously targeted residues used for modification of the regioselectivity of the enzyme. Finally, we have provided an example of a simple and elegant process for the detailed description of the network of cavities and tunnels, which can be used in the planning of enzyme modifications and can be easily adapted to the study of any other protein.
|
The modelling and enhancement of water hydrodynamics: general discussion. Baaden, M.; Borthakur, M. P.; Casanova, S.; Coalson, R.; Freger, V.; Gonzalez, M.; Góra, A.; Hinds, B.; Hirunpinyopas, W.; Hummer, G.; Kumar, M.; Lynch, C.; Murail, S.; Noy, A.; Sansom, M.; Song, Q.; Vashisth, H.; Vögele, M., Faraday Discuss. 2018, 354–360, doi:10.1039/C8FD90021C. ARTICLE |
||||||||
|
Structure and function of natural proteins for water transport: General discussion. Baaden, M.; Barboiu, M.; Bill, R. M.; Casanova, S.; Chen, C. L.; Conner, M.; Freger, V.; Gong, B.; Góra, A.; Hinds, B.; Horner, A.; Hummer, G.; Kumar, M.; Lokesh, M.; Mitra, S.; Noy, A.; Pohl, P.; Sadet, A.; Sansom, M.; Törnroth-Horsefield, S.; Vashisth, H., Faraday Discuss. 2018, 209, 83–95, doi:10.1039/C8FD90019A. ARTICLE |
||||||||
|
BALCONY: an R package for MSA and functional compartments of protein variability analysis. A. Płuciennik, M. Stolarczyk, M. Bzówka, A. Raczyńska, T. Magdziarz, A. Góra, BMC Bioinformatics 2018, 19: 300. DOI:10.1186/s12859-018-2294-z. ARTICLEOpen Access |
||||||||
ABSTRACT
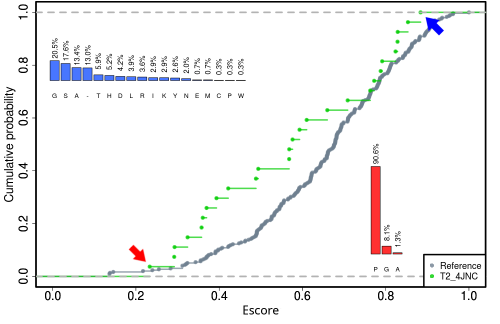 Background: Here, we present an R package for entropy/variability analysis that facilitates prompt and convenient data extraction, manipulation and visualization of protein features from multiple sequence alignments. BALCONY can work with residues dispersed across a protein sequence and map them on the corresponding alignment of homologous protein sequences. Additionally, it provides several entropy and variability scores that indicate the conservation of each residue.
Background: Here, we present an R package for entropy/variability analysis that facilitates prompt and convenient data extraction, manipulation and visualization of protein features from multiple sequence alignments. BALCONY can work with residues dispersed across a protein sequence and map them on the corresponding alignment of homologous protein sequences. Additionally, it provides several entropy and variability scores that indicate the conservation of each residue.
Results: Our package allows the user to visualize evolutionary variability by locating the positions most likely to vary and to assess mutation candidates in protein engineering.
Conclusion: In comparison to other R packages BALCONY allows conservation/variability analysis in context of protein structure with linkage of the appropriate metrics with physicochemical features of user choice.
Availability: CRAN project page: https://cran.r-project.org/package=BALCONY and our website: http://www.tunnelinggroup.pl/software/ for major platforms: Linux/Unix, Windows and Mac OS X.
|
Modulating D-amino acid oxidase (DAAO) substrate specificity through facilitated solvent access. K. Subramanian, A. Góra, R. Spruijt, K. Mitusińska, M. Suarez-Diez, V. M. dos Santos, P. J. Schaap, PLoS ONE 13(6): e0198990. DOI: 10.1371/journal.pone.0198990 ARTICLEOpen Access |
||||||||
ABSTRACT
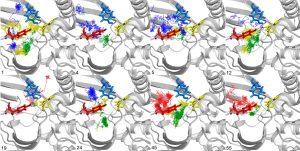
D-amino acid oxidase (DAAO) degrades D-amino acids to produce α-ketoacids, hydrogen peroxide and ammonia. DAAO has often been investigated and engineered for industrial and clinical applications. We combined information from literature with a detailed analysis of the structure to engineer mammalian DAAOs. The structural analysis was complemented with molecular dynamics simulations to characterize solvent accessibility and product release mechanisms. We identified non-obvious residues located on the loops on the border between the active site and the secondary binding pocket essential for pig and human DAAO substrate specificity and activity. We engineered DAAOs by mutating such critical residues and characterised the biochemical activity of the resulting variants. The results highlight the importance of the selected residues in modulating substrate specificity, product egress and enzyme activity, suggesting further steps of DAAO re-engineering towards desired clinical and industrial applications.
2017
|
AQUA-DUCT a ligands tracking tool. T. Magdziarz, K. Mitusińska, S. Gołdowska, A. Płuciennik, M. Stolarczyk, M .Ługowska and A. Góra; Bioinformatics (2017) 33 (13): 2045-2046. DOI: 10.1093/bioinformatics/btx125 ARTICLE or PROOF VERSIONOpen Access |
||||||||
ABSTRACT
Motivation:The identification and tracking of molecules which enter active site cavity requires screening the positions of thousands of single molecules along several thousand molecular dynamic steps. To fill the existing gap between tools searching for tunnels and pathways and advanced tools employed for accelerated water flux investigations, we have developed AQUA-DUCT.
Results: AQUA-DUCT is an easy-to-use tool that facilitates analysis of the behaviour of molecules that penetrate any selected region in a protein. It can be used for any type of molecules e.g., water, oxygen, carbon dioxide, organic solvents, ions.
Platform and Availability: Linux, Windows, macOS, OpenBSD, http://www.aquaduct.pl
Contact: a.gora@tunnelinggroup.pl, info@aquaduct.pl
The available copy of paper is a pre-copyedited, author-produced version of an article accepted for publication in Bioinformatics following peer review. The version of record Bioinformatics (2017) 33 (13): 2045-2046. is available online at: https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btx125/3059930/AQUA-DUCT-a-ligands-tracking-tool?redirectedFrom=fulltext DOI: 10.1093/bioinformatics/btx125.
2016
|
Engineering a de Novo Transport Tunnel ACS J. Brezovsky, P. Babkova, O. Degtjarik, A. Fortova, A. Gora, I. Iermak, P. Rezacova, P. Dvorak, I. Smatanova, Z. Prokop, R. Chaloupkova, and J. Damborsky, Catalysis 2016, 6, 7597-7610 DOI: 10.1021/acscatal.6b02081 ARTICLE |
||||||||
ABSTRACT
Transport of ligands between buried active sites and bulk solvent is a key step in the catalytic cycle of many enzymes. Absence of evolutionary optimized transport tunnels is an important barrier limiting the efficiency of biocatalysts prepared by computational design. Creating a structurally defined and functional “hole” into the protein represents an engineering challenge. Here we describe the computational design and directed evolution of a de novo transport tunnel in haloalkane dehalogenase. Mutants with a blocked native tunnel and newly opened auxiliary tunnel in a distinct part of the structure showed dramatically modified properties. The mutants with blocked tunnels acquired specificity never observed with native family members, up to 32-times increased substrate inhibition and 17-times reduced catalytic rates. Opening of the auxiliary tunnel resulted in specificity and substrate inhibition similar to the native enzyme, and the most proficient haloalkane dehalogenase reported to date (kcat = 57 s-1 with 1,2-dibromoethane at 37oC and pH=8.6). Crystallographic analysis and molecular dynamics simulations confirmed successful introduction of structurally defined and functional transport tunnel. Our study demonstrates that whereas we can open the transport tunnels with reasonable proficiency, we cannot accurately predict the effects of such change on the catalytic properties. We propose that one way to increase efficiency of an enzyme is the direct its substrates and products into spatially distinct tunnels. The results clearly show the benefits of enzymes with de novo transport tunnels and we anticipate that this engineering strategy will facilitate creation of a wide range of useful biocatalysts.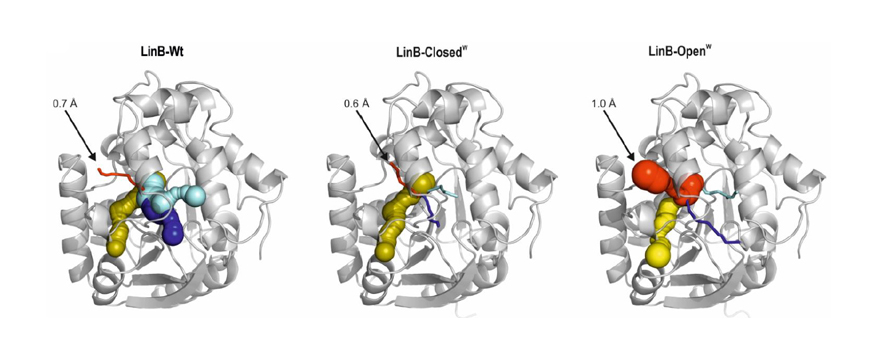
|
Molecular descriptor data explain market prices of a large commercial chemical compound library J. Polanski, U. Kucia, R. Duszkiewicz, A. Kurczyk, T. Magdziarz and J. Gasteiger, Scientific Reports 6, Article number: 28521 (2016). doi:10.1038/srep28521 ARTICLEOpen Access |
||||||||
ABSTRACT
The relationship between the structure and a property of a chemical compound is an essential concept in chemistry  guiding, for example, drug design. Actually, however, we need economic considerations to fully understand the fate of drugs on the market. We are performing here for the first time the exploration of quantitative structure-economy relationships (QSER) for a large dataset of a commercial building block library of over 2.2 million chemicals. This investigation provided molecular statistics that shows that on average what we are paying for is the quantity of matter. On the other side, the influence of synthetic availability scores is also revealed. Finally, we are buying substances by looking at the molecular graphs or molecular formulas. Thus, those molecules that have a higher number of atoms look more attractive and are, on average, also more expensive. Our study shows how data binning could be used as an informative method when analyzing big data in chemistry.
guiding, for example, drug design. Actually, however, we need economic considerations to fully understand the fate of drugs on the market. We are performing here for the first time the exploration of quantitative structure-economy relationships (QSER) for a large dataset of a commercial building block library of over 2.2 million chemicals. This investigation provided molecular statistics that shows that on average what we are paying for is the quantity of matter. On the other side, the influence of synthetic availability scores is also revealed. Finally, we are buying substances by looking at the molecular graphs or molecular formulas. Thus, those molecules that have a higher number of atoms look more attractive and are, on average, also more expensive. Our study shows how data binning could be used as an informative method when analyzing big data in chemistry.
PUBLICATIONS BEFORE TUNNELING GROUP ESTABLISHMENT
|
CAVER Analyst 1.0: graphic tool for interactive visualization and analysis of tunnels and channels in protein structures B. Kozlikova, E. Sebestova, V. Sustr, J. Brezovsky, O. Strnad, L. Daniel, D. Bednar, A. Pavelka, M. Manak, M. Bezdeka, P. Benes, M. Kotry, A. Gora, J. Damborsky, and J. Sochor, Bioinforma. Oxf. Engl., vol. 30, no. 18, pp. 2684–2685, Sep. 2014 ARTICLE. |
||||||||
ABSTRACT
The transport of ligands, ions or solvent molecules into proteins with buried binding sites or through the membrane is enabled by protein tunnels and channels. CAVER Analyst is a software tool for calculation, analysis and real-time visualization of access tunnels and channels in static and dynamic protein structures. It provides an intuitive graphic user interface for setting up the calculation and interactive exploration of identified tunnels/channels and their characteristics.
|
Gates of Enzymes A. Gora, J. Brezovsky, and J. Damborsky, Chem. Rev., vol. 113, no. 8, pp. 5871–5923, 2013. ARTICLEOpen Access |
||||||||
Table of Contents
- Introduction
- Molecular Function of Gates
- Structural Basis of Gates
- Locations of Gates
- Engineering of Gates
- Conclusions
ABSTRACT
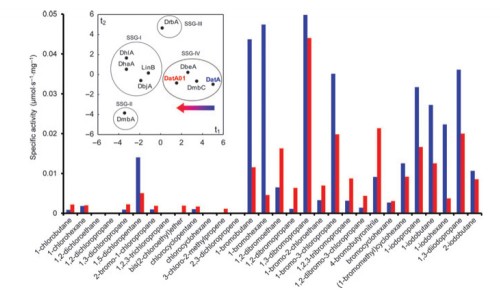 Haloalkane dehalogenases catalyse the hydrolysis of carbon-halogen bonds in various chlorinated, brominated and iodinated compounds. These enzymes have a conserved pair of halide-stabilising residues that are important in substrate binding and stabilisation of the transition state and the halide ion product via hydrogen bonding. In all previously known haloalkane dehalogenase, these residues are either a pair of tryptophans or a tryptophan-asparagine pair. The newly isolated haloalkane dehalogenase DatA from Agrobacterium tumefaciens C58 possesses a unique halide-stabilising tyrosine residue, Y109, in place of the conventional tryptophan. A variant of DatA with the Y109W mutation was created and the effects of this mutation on the enzyme’s structure and catalytic properties were studied using spectroscopy and pre-steady-state kinetic experiments. Quantum mechanical and molecular dynamics calculations were used to obtain a detailed analysis of the hydrogen bonding patterns within the active sites of the wild-type and the mutant, and of the stabilisation of the ligands as the reaction proceeds. Fluorescence quenching experiments suggested that replacing the tyrosine with tryptophan improves halide binding 3.7-fold, presumably due to the introduction of an additional hydrogen bond. Kinetic analysis revealed that the mutation affected the enzyme’s substrate specificity and reduced its K0.5 for selected halogenated substrates by a factor of 2-4, without impacting the rate-determining hydrolytic step. We conclude that DatA is the first natural haloalkane dehalogenase that stabilises its substrate in the active site using only a single hydrogen bond, which is a new paradigm in catalysis by this enzyme family.
Haloalkane dehalogenases catalyse the hydrolysis of carbon-halogen bonds in various chlorinated, brominated and iodinated compounds. These enzymes have a conserved pair of halide-stabilising residues that are important in substrate binding and stabilisation of the transition state and the halide ion product via hydrogen bonding. In all previously known haloalkane dehalogenase, these residues are either a pair of tryptophans or a tryptophan-asparagine pair. The newly isolated haloalkane dehalogenase DatA from Agrobacterium tumefaciens C58 possesses a unique halide-stabilising tyrosine residue, Y109, in place of the conventional tryptophan. A variant of DatA with the Y109W mutation was created and the effects of this mutation on the enzyme’s structure and catalytic properties were studied using spectroscopy and pre-steady-state kinetic experiments. Quantum mechanical and molecular dynamics calculations were used to obtain a detailed analysis of the hydrogen bonding patterns within the active sites of the wild-type and the mutant, and of the stabilisation of the ligands as the reaction proceeds. Fluorescence quenching experiments suggested that replacing the tyrosine with tryptophan improves halide binding 3.7-fold, presumably due to the introduction of an additional hydrogen bond. Kinetic analysis revealed that the mutation affected the enzyme’s substrate specificity and reduced its K0.5 for selected halogenated substrates by a factor of 2-4, without impacting the rate-determining hydrolytic step. We conclude that DatA is the first natural haloalkane dehalogenase that stabilises its substrate in the active site using only a single hydrogen bond, which is a new paradigm in catalysis by this enzyme family.
|
Software tools for identification, visualization and analysis of protein tunnels and channels J. Brezovsky, E. Chovancova, A. Gora, A. Pavelka, L. Biedermannova, and J. Damborsky, Biotechnol. Adv., vol. 31, no. 1, pp. 38–49, Jan. 2013. ARTICLE |
||||||||
ABSTRACT
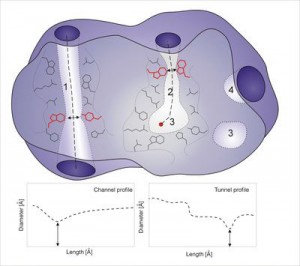 Protein structures contain highly complex systems of voids, making up specific features such as surface clefts or grooves, pockets, protrusions, cavities, pores or channels, and tunnels. Many of them are essential for the migration of solvents, ions and small molecules through proteins, and their binding to the functional sites. Analysis of these structural features is very important for understanding of structure-function relationships, for the design of potential inhibitors or proteins with improved functional properties. Here we critically review existing software tools specialized in rapid identification, visualization, analysis and design of protein tunnels and channels. The strengths and weaknesses of individual tools are reported together with examples of their applications for the analysis and engineering of various biological systems. This review can assist users with selecting a proper software tool for study of their biological problem as well as highlighting possible avenues for further development of existing tools. Development of novel descriptors representing not only geometry, but also electrostatics, hydrophobicity or dynamics, is needed for reliable identification of biologically relevant tunnels and channels.
Protein structures contain highly complex systems of voids, making up specific features such as surface clefts or grooves, pockets, protrusions, cavities, pores or channels, and tunnels. Many of them are essential for the migration of solvents, ions and small molecules through proteins, and their binding to the functional sites. Analysis of these structural features is very important for understanding of structure-function relationships, for the design of potential inhibitors or proteins with improved functional properties. Here we critically review existing software tools specialized in rapid identification, visualization, analysis and design of protein tunnels and channels. The strengths and weaknesses of individual tools are reported together with examples of their applications for the analysis and engineering of various biological systems. This review can assist users with selecting a proper software tool for study of their biological problem as well as highlighting possible avenues for further development of existing tools. Development of novel descriptors representing not only geometry, but also electrostatics, hydrophobicity or dynamics, is needed for reliable identification of biologically relevant tunnels and channels.
|
A single mutation in a tunnel to the active site changes the mechanism and kinetics of product release in haloalkane dehalogenase LinB L. Biedermannová, Z. Prokop, A. Gora, E. Chovancová, M. Kovács, J. Damborsky, and R. C. Wade, J. Biol. Chem., vol. 287, no. 34, pp. 29062–29074, Aug. 2012.ARTICLE |
||||||||
ABSTRACT
Many enzymes have buried active sites. The properties of the tunnels connecting the active site with bulk solvent affect ligand binding and unbinding and also the catalytic properties. Here, we investigate ligand passage in the haloalkane dehalogenase enzyme LinB and the effect of replacing leucine by a bulky tryptophan at a tunnel-lining position. Transient kinetic experiments show that the mutation significantly 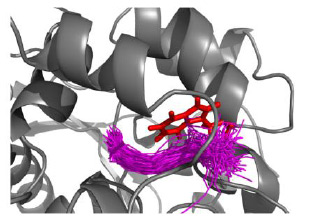 slows down the rate of product release. Moreover, the mechanism of bromide ion release is changed from a one-step process in the wild type enzyme to a two-step process in the mutant. The rate constant of bromide ion release corresponds to the overall steady-state turnover rate constant, suggesting that product release became the rate-limiting step of catalysis in the mutant. We explain the experimental findings by investigating the molecular details of the process computationally. Analysis of trajectories from molecular dynamics simulations with a tunnel detection software reveals differences in the tunnels available for ligand egress. Corresponding differences are seen in simulations of product egress using a specialized enhanced sampling technique. The differences in the free energy barriers for egress of a bromide ion obtained using potential of mean force calculations are in good agreement with the differences in rates obtained from the transient kinetic experiments. Interactions of the bromide ion with the introduced tryptophan are shown to affect the free energy barrier for its passage. The study demonstrates how the mechanism of an enzymatic catalytic cycle and reaction kinetics can be engineered by modification of protein tunnels.
slows down the rate of product release. Moreover, the mechanism of bromide ion release is changed from a one-step process in the wild type enzyme to a two-step process in the mutant. The rate constant of bromide ion release corresponds to the overall steady-state turnover rate constant, suggesting that product release became the rate-limiting step of catalysis in the mutant. We explain the experimental findings by investigating the molecular details of the process computationally. Analysis of trajectories from molecular dynamics simulations with a tunnel detection software reveals differences in the tunnels available for ligand egress. Corresponding differences are seen in simulations of product egress using a specialized enhanced sampling technique. The differences in the free energy barriers for egress of a bromide ion obtained using potential of mean force calculations are in good agreement with the differences in rates obtained from the transient kinetic experiments. Interactions of the bromide ion with the introduced tryptophan are shown to affect the free energy barrier for its passage. The study demonstrates how the mechanism of an enzymatic catalytic cycle and reaction kinetics can be engineered by modification of protein tunnels.
|
CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures E. Chovancova, A. Pavelka, P. Benes, O. Strnad, J. Brezovsky, B. Kozlikova, A. Gora, V. Sustr, M. Klvana, P. Medek, L. Biedermannova, J. Sochor, and J. Damborsky, PLoS Comput Biol, vol. 8, no. 10, p. e1002708, Oct. 2012.ARTICLEOpen Access |
||||||||
ABSTRACT
Tunnels and channels facilitate the transport of small molecules, ions and water solvent in a large variety of proteins. Characteristics of individual transport pathways, including their geometry, physico-chemical properties and dynamics are instrumental for understanding of structure-function relationships of these proteins, for the design of new inhibitors and construction of improved biocatalysts. CAVER is a software tool widely used for the identification and characterization of transport pathways in static macromolecular structures.
 Herein we present a new version of CAVER enabling automatic analysis of tunnels and channels in large ensembles of protein conformations. CAVER 3.0 implements new algorithms for the calculation and clustering of pathways. A trajectory from a molecular dynamics simulation serves as the typical input, while detailed characteristics and summary statistics of the time evolution of individual pathways are provided in the outputs. To illustrate the capabilities of CAVER 3.0, the tool was applied for the analysis of molecular dynamics simulation of the microbial enzyme haloalkane dehalogenase DhaA. CAVER 3.0 safely identified and reliably estimated the importance of all previously published DhaA tunnels, including the tunnels closed in DhaA crystal structures. Obtained results clearly demonstrate that analysis of molecular dynamics simulation is essential for the estimation of pathway characteristics and elucidation of the structural basis of the tunnel gating. CAVER 3.0 paves the way for the study of important biochemical phenomena in the area of molecular transport, molecular recognition and enzymatic catalysis. The software is freely available as a multiplatform command-line application at http://www.caver.cz.
Herein we present a new version of CAVER enabling automatic analysis of tunnels and channels in large ensembles of protein conformations. CAVER 3.0 implements new algorithms for the calculation and clustering of pathways. A trajectory from a molecular dynamics simulation serves as the typical input, while detailed characteristics and summary statistics of the time evolution of individual pathways are provided in the outputs. To illustrate the capabilities of CAVER 3.0, the tool was applied for the analysis of molecular dynamics simulation of the microbial enzyme haloalkane dehalogenase DhaA. CAVER 3.0 safely identified and reliably estimated the importance of all previously published DhaA tunnels, including the tunnels closed in DhaA crystal structures. Obtained results clearly demonstrate that analysis of molecular dynamics simulation is essential for the estimation of pathway characteristics and elucidation of the structural basis of the tunnel gating. CAVER 3.0 paves the way for the study of important biochemical phenomena in the area of molecular transport, molecular recognition and enzymatic catalysis. The software is freely available as a multiplatform command-line application at http://www.caver.cz.
PREECIDINGS AND OTHER PAPERS
Gora, A., Brezovsky, J. & Damborsky, J., Computer-assisted enzyme engineering by modification of tunnels, channels and gates. Current Opinion in Biotechnology vol 22, supplement 1 September 2011.
Gora, A., Pacheco Tanaka, D. A., Mizukami, F. & Suzuki, T. M., Low temperature hydrogen recovering from organic storage compounds – application of pore fill type palladium membrane. Proceedings of International Conference and Exhibition on Green Chemistry, 18-21 IX 2006 Kuala Lumpur, Malaysia, 2006.
Gora, A. & Brocławik E. Dissociation of the Water Molecule on the V-W-O Catalyst Surface – Quantum Chemical Modeling. Polish Journal of Environmental Studies, 9(1), 31-34, 2000.
Najbar M., Białas A., Mizukami F., Wesełucha-Birczyńska A., Bielańska E. & Gora A. Vanadia-Tungsta DENOX Catalysts on High Surface Area Rutile. Polish Journal of Environmental Studies, 6, 83-8, 1997.
BOOKS
Prokop Z, Gora A, Brezovsky J, Chalupkova R, Stepankova V & Damborsky J Protein Engineering Handbook, Volume 3: Book chapter: Engineering of protein tunnels: Keyhole-lock-key model for catalysis by the enzymes with buried active sites – ISBN 978-3-527-33123-9 – Wiley-VCH, Weinheim.
PATENTS
US Patent No 13/604,094 “Method of thermostabilization of a protein and/or stabilization towards organic solvents” Jiri Damborsky, Zbynek Prokop, Tana Koudelakova, Veronika Stepankova, Radka Chaloupkova, Eva Chovancova, Artur Wiktor Gora, Jan Brezovsky.